
Best practices for nonadiabatic molecular dynamics simulations [Article v1.0]
DOI:
https://doi.org/10.33011/livecoms.7.1.4157Abstract
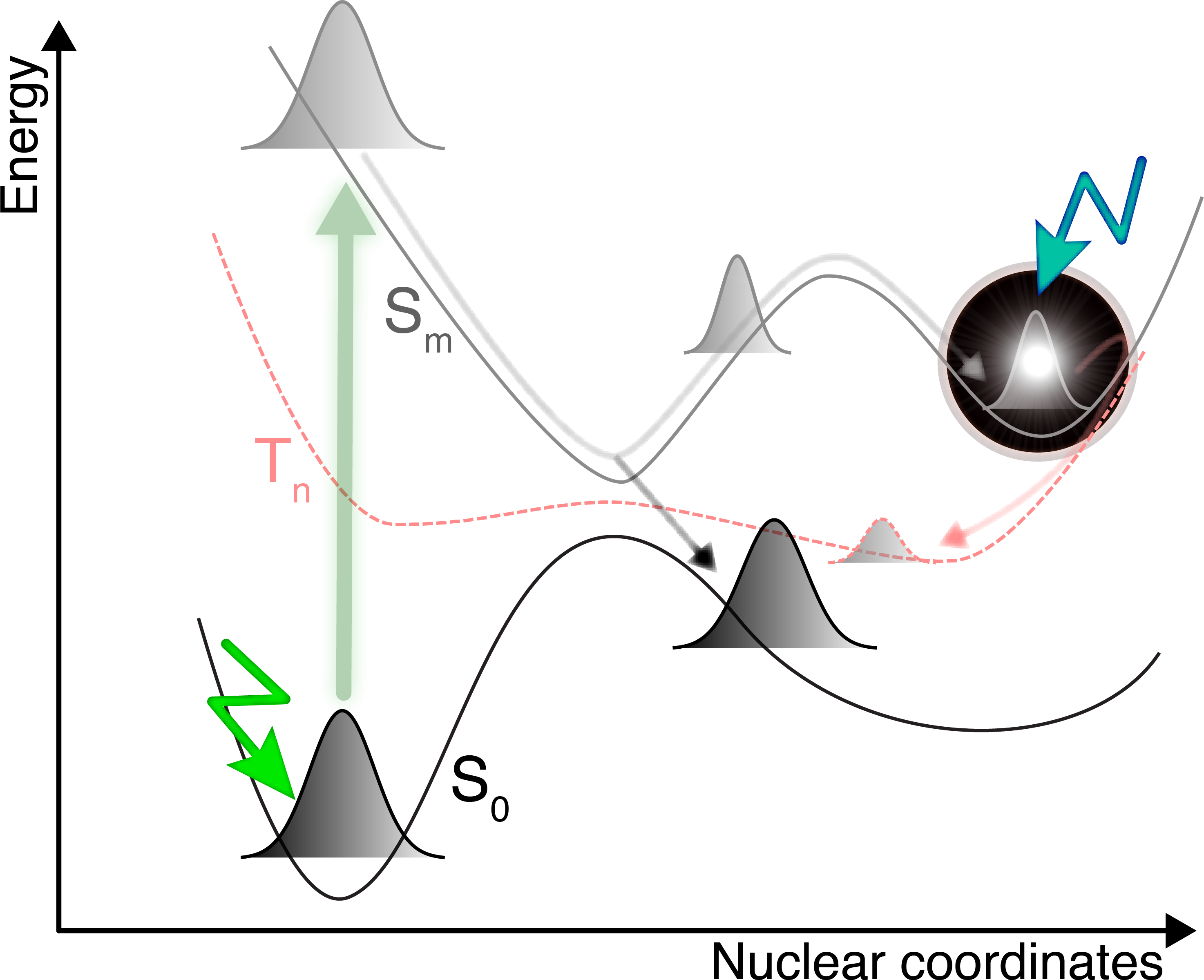
Nonadiabatic molecular dynamics simulations aim to describe the coupled electron- nuclear dynamics of molecules in excited electronic states, beyond the celebrated Born-Oppenheimer approximation. These simulations have been applied to understand a plethora of photochemical and photophysical processes and to support the interpretation of ultrafast spectroscopy experiments at advanced light sources. As a result, the number of nonadiabatic dynamics simulations has been growing significantly over the past decade. Yet, the field remains in its infancy, and a potential user may find it difficult to approach this type of simulation, given their complexity and the number of elements that should be considered for a (hopefully) successful nonadiabatic dynamics simulation. Nonadiabatic molecular dynamics relies on several key steps: finding a level of electronic-structure theory to describe the molecule in its Franck-Condon region and beyond, describing the photoexcitation process, selecting a method to perform the nonadiabatic dynamics, and analyzing the final results before calculating observables for a more direct comparison with experiment. This Best Practices guide aims to provide a general guide for the user of nonadiabatic molecular dynamics by (i) discussing the fundamentals of nonadiabatic molecular dynamics and the various trajectory-based methods developed for molecular systems, (ii) introducing the different electronic-structure methods and concepts – adiabatic/diabatic representation, conical intersections – that can be used with nonadiabatic molecular dynamics (or for benchmarking), (iii) providing details on the various steps required to perform a nonadiabatic dynamics simulation and their practical use, as well as guided examples and a discussion on the calculation of observables, (iv) proposing a FAQ with the typical questions a user may have when performing nonadiabatic dynamics, and (v) sketching a checklist for the key practical steps when performing a (trajectory-based) nonadiabatic molecular dynamics. Each section is self-contained, but we endeavor to provide additional key references for each concept discussed, making this Guide a starting point for the interested reader to dig further into the field of nonadiabatic dynamics.
Downloads
Published
How to Cite
License
Copyright (c) 2026 Antonio Prlj, Jack T. Taylor, Jiří Janoš, Elise Lognon, Daniel Hollas, Petr Slavíček, Federica Agostini, Basile F. E. Curchod

This work is licensed under a Creative Commons Attribution 4.0 International License.
