
Running Gaussian-accelerated Molecular Dynamics Simulations in NAMD [Article v1.0]
DOI:
https://doi.org/10.33011/livecoms.6.1.3815Keywords:
Enhanced sampling, Molecular dynamics simulations, Free energy surfacesAbstract
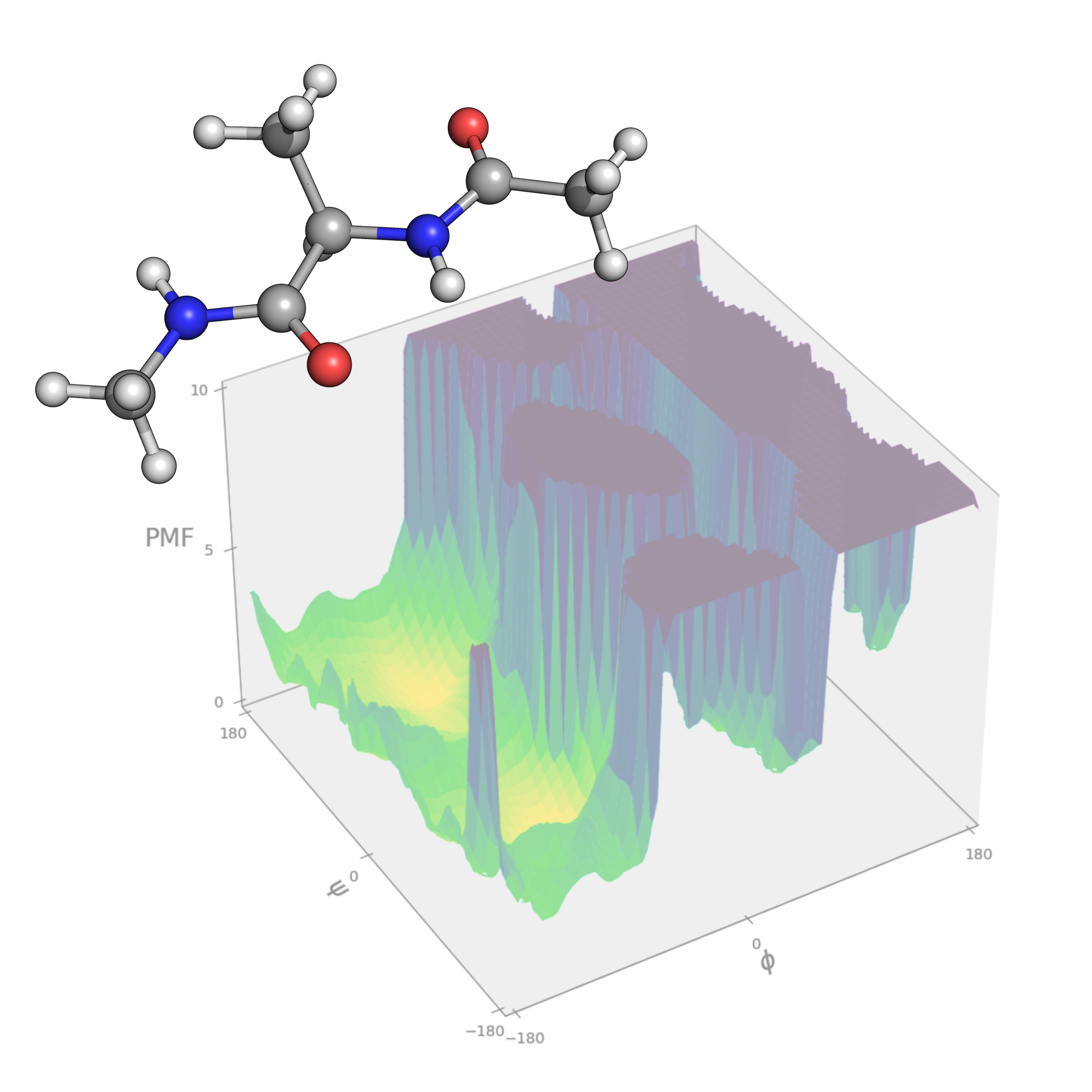
Gaussian-accelerated molecular dynamics (GaMD) simulations are an advanced technique that enhances the sampling of configurational space by applying biasing potentials that reduce energy barriers, enabling faster exploration of the free energy landscape. This tutorial demonstrates the application of GaMD to the alanine dipeptide, serving as an accessible model system, and guides users through all GaMD simulation stages: conventional MD, GaMD equilibration, GaMD production, and reweighting. Users will gain practical insights into the preparation of input files, monitoring of GaMD convergence, and analysis of free energy profiles using PyReweighting.
We make a particular effort to connect the underlying theory with the GaMD workflow. This tutorial is intended for users with prior molecular dynamics experience, Linux and command-line navigation, and with basic Python knowledge. The step-by-step instructions and accompanying scripts aim to streamline the GaMD workflow, making it accessible for the broader research community to explore enhanced sampling for a range of biomolecular systems.
Downloads
Published
How to Cite
License
Copyright (c) 2025 Haley M. Michel, Marcelo D. Polêto, Justin A. Lemkul

This work is licensed under a Creative Commons Attribution 4.0 International License.
