
Computing Absolute Binding Affinities by Streamlined Alchemical Free Energy Perturbation (SAFEP) [Article v1.0]
DOI:
https://doi.org/10.33011/livecoms.5.1.2067Keywords:
absolute binding affinity, free energy, alchemyAbstract
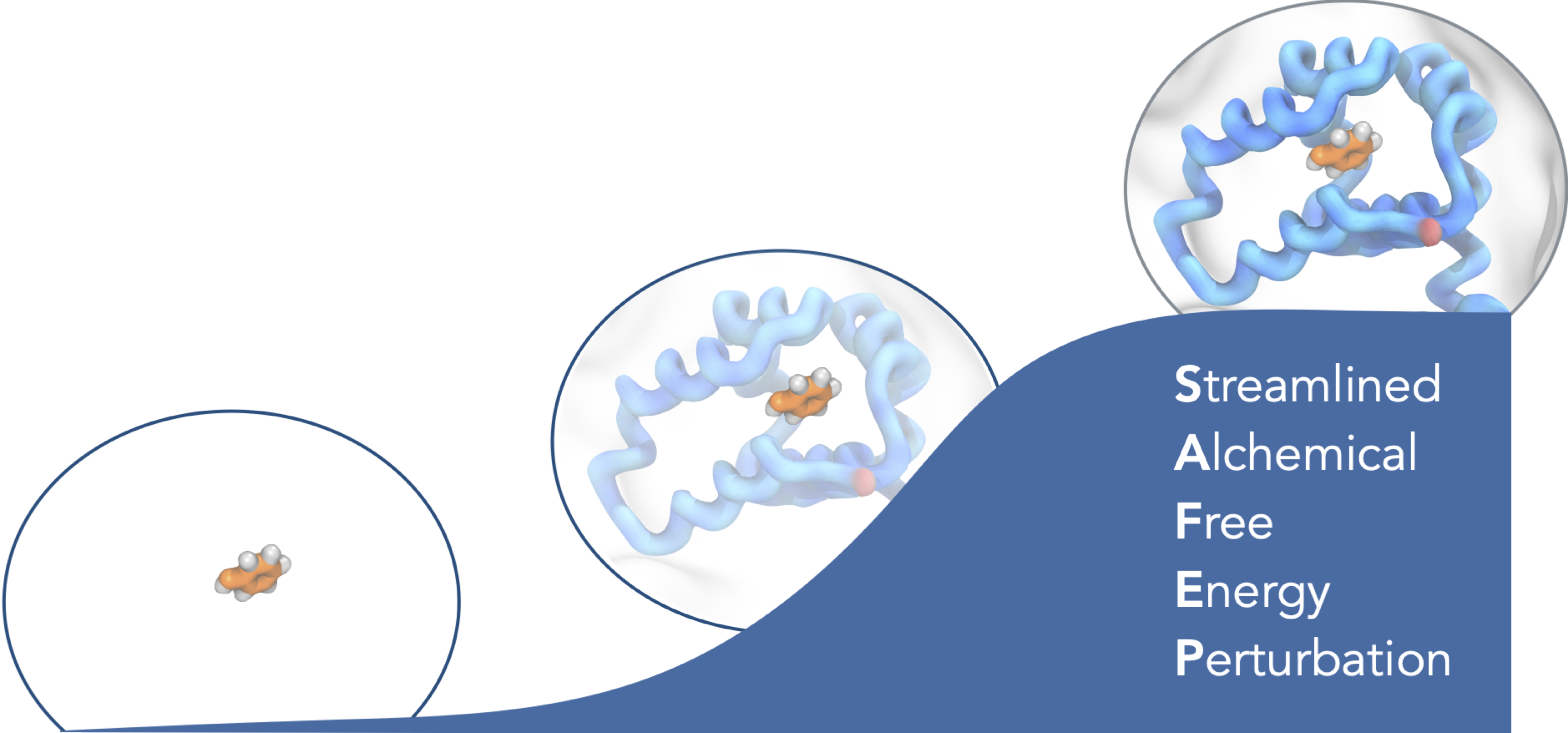
Free Energy Perturbation (FEP) is a powerful but challenging computational technique for estimating differences in free energy between two or more states. This document is intended both as a tutorial and as an adaptable protocol for computing free energies of binding using free energy perturbations in NAMD. We present the Streamlined Alchemical Free Energy Perturbation (SAFEP) framework. SAFEP shifts the computational frame of reference from the ligand to the binding site itself. This both simplifies the thermodynamic cycle and makes the approach more broadly applicable to superficial sites and other less common geometries. As a practical example, we give instructions for calculating the absolute binding free energy of phenol to lysozyme. We assume familiarity with standard procedures for setting up, running, and analyzing molecular dynamics simulations using NAMD and VMD. While simulation times will vary, the human tasks should take no more than 3 to 4 hours for a reader without previous training in free energy calculations or ex- perience with the VMD Colvars Dashboard. Sample data are provided for all key calculations both for comparison and readers’ convenience.
Downloads
Published
How to Cite
License
Copyright (c) 2023 Ezry Santiago-McRae, Mina Ebrahimi, Jesse W. Sandberg, Grace Brannigan, Jérôme Hénin

This work is licensed under a Creative Commons Attribution 4.0 International License.
